Blog, Discovering New Metalloantibiotics with Machine Learning
Hi there, Angelo here. We recently published our first paper applying Machine Learning (ML) to the discovery of metalloantibiotics. The Paper is accessible here (Journal Website) or as a Preprint
Here I wanted to provide a little bit more background on how this work came to be, what I think it means and where we are going next.
Background
I started getting into ML during the long Lockdowns of the pandemic. First it was a fun side project but after a while I realised that this had interesting applications in the field of chemistry in general and could specifically also find uses in our concrete field of metalloantibiotic discovery. The bread and butter of any ML-project is data to train your models on. Unfortunately when it comes to both metal complexes and biological assays, there is a) not much or no good data out there and biological data is notorious for being quite noisy when it is collated from across different labs. The first step was therefore to generate our own, high-quality data. It just so happened that I had started a collaboration with Prof. Wee Han Ang at NUS in Singapore where we explored the antibacterial properties of 288 combinatorially prepared ruthenium complexes. These compounds had all been synthesized and tested under identical laboratory conditions providing a small but good quality dataset.
The next issue was that in order to do ML projects with molecules, the latter need to be somehow translated into something a computer can understand.
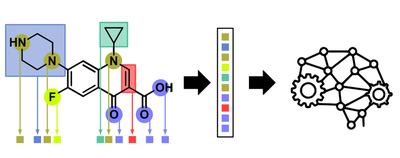
Unfortunately SMILES strings cannot easily be generated for molecules containing transition metals as these do not follow covalency rules as nicely as purely organic molecules. So we had to use a trick. Because all 288 ruthenium complexes in our first dataset contained the same metal (ruthenium) in the same oxidation state (+2) this meant that the only difference inbetween two given complexes was the nature of the Schiff-base and Arene ligands. We therefore hypothesized that it would be sufficient to describe a given metal complex as a linear combination of its ligand components (effectively ‘bracketing out’ the metal).
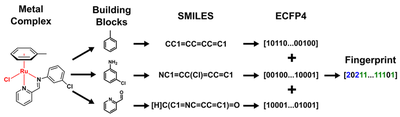
Next we took the antibacterial activity data and converted it into a binary format: 1 if the specific complex was active against the tested bacterial strain (MRSA) and 0 if it was not active. Now we have a feature vector and target variable for all 288 molecules and we could start training ML algorithms. Because this was a relatively simple ML project we opted for ‘out-of-the-box’ ML models using the popular scikit-learn library. Because our dataset was highly imbalanced i.e. only a small number of compounds were active versus a lot of inactive ones, we had to be very careful to not end up with a model that just predicts everything to be inactive (where it would still be right 90& of the time). We checked this by a) scrambling the data and showing that if labels are assigned randomly the learning is not better than random and b) by looking at the confusion matrices and checking that both actives are predicted to be active and inactives are correctly predicted to be inactive. Overall the learning worked really well with almost all standard ML models. However up to this point we just showed that we can train a ML model on antibacterial data of some metal complexes… not really a big deal because we have not done anything with this model yet. So we wanted to validate that this model is actually able to predict the activity of complexes nobody has ever made before. To do this we put together a library of over 77 million possible ruthenium arene Schiff-base complexes (based on commercially available building blocks) and showed them to our best ML models. These predicted around 2 million of these to be active. We then had a closer look at these 2 million, identifying building blocks that appeared most frequently and narrowing it down to ones that are actually commercially available and affordable. A few months later and about 1200 USD poorer the building blocks had arrived. The Ang group went to work and prepared a new combinatorial library of 54 complexes and tested them against bacteria.
Now it is worth revisiting what we had done up to this point: We had taken a library of 288 compounds that were tested, converted those metal complexes into something we thought made sense to a computer and used that to train ML models able to predict if a given ruthenium complex is active against bacteria. We then came up with many millions of hypothetical compounds we could make and our model tossed out everything it predicted not to be active. Finally we narrowed it down and ordered the real chemical components to make 54 compounds based on the predictions of a computer. Safe to say that I was quite nervous about the results because a small error somewhere in the code would have meant that the ML model’s predictions were useless.

Some thoughts
I am very pleased on how this turned out and that we got a very nice paper out of it. But I view this very much as a proof of concept study showing that we can use ML in our workflow. It shows that we can approximate metal complexes of a given class by their ligands, and that the ML predictions can be translated in the lab to actual results. But there are also several limitations to this work. Firstly it is limited to a specific class of metal complexes. Secondly active/inactive against a specific strain is not a super useful metric because it a) does not tell us how active a given compound will be and it b) also does not tell us anything about other very important properties such as e.g. toxicity. The good news is, we can work on that now!
Outlook
We are currently working on several aspects of this to drive this further. This means first and foremost: More and better data! This will allow us to train better and more high-resolution models able to not only predict if a given compound will be active but also how strongly it will be active. Also we are aiming to include more information such as toxicity as well.
Secondly we are working to develop ways to describe metal complexes for computational approaches which do not limit us to a single compound class. We are close to releasing a preprint on our exciting efforts in this space so stay tuned!
Overall I am very excited about the possibilities in this space. If you are interested in working with us on expanding the use of ML in the field of metalloantibiotics or beyond, please do reach out, we are keen to collaborate and share!